02 Pulmonary2
Mesothelioma is a malignant tumor of the visceral or parietal pleura.
It results from long-term exposure to asbestos.
Smoking does NOT increase the risk of mesothelioma in asbestos workers. This is in contrast to the synergistic effect of smoking and asbestos exposure on developing primary bronchogenic lung carcinoma.
The most common presenting symptoms of **mesothelioma **are chest pain, dyspnea, and recurrent idiopathic pleural effusions.
Diagnosis of malignant mesothelioma requires tissue biopsy.
Chest x-ray will show pleural thickening and possibly pleural effusion.
Aggressive therapy including combined pneumonectomy, chemotherapy and radiation has been shown to improve outcomes of patients with epithelioid type mesothelioma.
Malignant mesothelioma has a poor prognosis. Mean survival time is approximately 12 months.
Pleurisy
Pleurisy, also known as pleuritis, is inflammation of the pleural lining of the lungs.
Pleurisy is commonly caused by:
- **Viral infections **(most common)
- Connective tissue disorders (e.g. systemic lupus erythematosus, rheumatoid arthritis)
- Bacterial infections (e.g. tuberculosis)
- Hypersensitivity pneumonitis
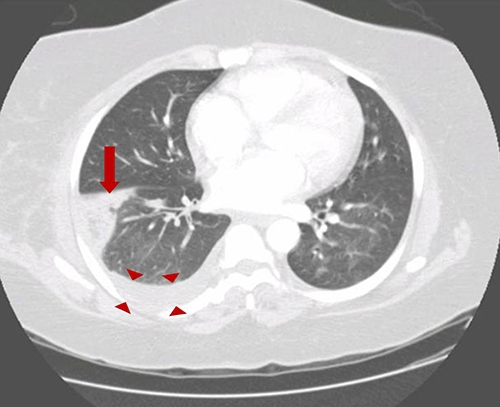
- Pulmonary emboli: Approximately 10% of patients with PE have occlusion of a peripheral pulmonary artery by thrombus, causing pulmonary infarction. These small peripheral thrombi are more likely to cause pleuritic chest pain and hemoptysis, due to inflammation and irritation of the lung parenchyma and adjacent visceral and parietal pleura. CT shows wedge shaped lungs and pleural effusion:
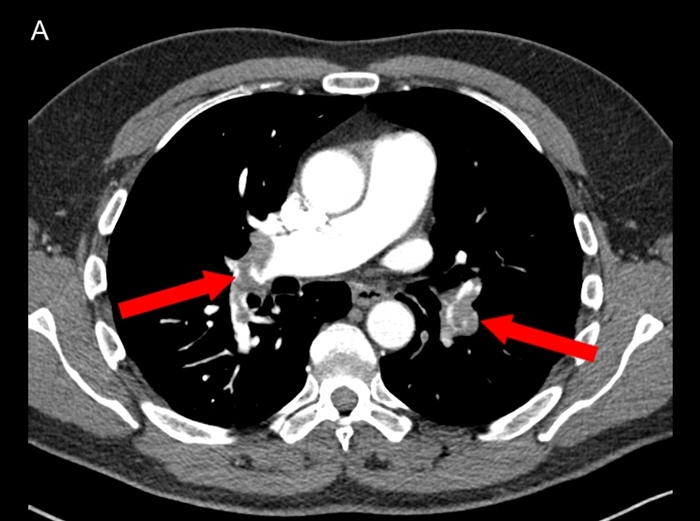
- contrast CT: pulmonary artery filling defect:
Patients with pleurisy typically present with:
- Intense, knife-like pain worse on inspiration
- Tachycardia or tachypnea (may be secondary to pain)
- Pleural friction rub on auscultation (sounds like Velcro)
- History of upper respiratory infection
The diagnosis of pleurisy is made by clinical exam and a chest x-ray to rule out serious etiologies.
The management of pleurisy includes:
- **Supportive therapy **(e.g. supplemental oxygen, bed rest)
- NSAIDs
Pulmonary abscess
Pulmonary abscess is a collection of puswithin the lung parenchyma with confined cavitation that results from a pulmonary infection.
The three bacterial causes of pulmonary abscesses are:
- Staphylococcus aureus in IV drug users with endocarditis.
- Klebsiella pneumoniae in obtunded alcoholics.
- Anaerobic oral flora in patients with aspiration pneumonia.
Bacteria from the oropharynx implicated in pulmonary abscesses from aspiration include:
- Bacteroides
- Fusobacterium
- Peptococcus
Pulmonary abscesses due to septic embolization should always be considered in patients with endocarditis and multiple cavitary pulmonary infiltrates.
Suspect pulmonary abscess in a patient who is at risk for aspiration who presents with:
- Productive cough
- Poor dentition
- Spiking fevers
Patients with pulmonary abscesses will often be predisposed to aspiration pneumonia due to** impaired epiglottic function** and therefore inadequate airway protection. Commonly tested scenarios include:
- Obtunded alcoholic
- Patient with seizure disorder
- Advanced dementia
Foul smelling sputum in a patient with aspiration is indicative of anaerobic infection.
Lung abscesses appear as solitary, cavitating lesions with an air-fluid level and surrounding infiltrateon CXR.
The superior segment of the right lower lobe is the most common site for aspiration. Therefore, many abscesses are often identified in the right lower lobe.
Unlike empyemas, lung abscess are treated for several months withempiric antibiotic therapy and do NOT require assisted drainage or surgery.
As pulmonary abscesses are often polymicrobial it is imperative that any antibiotic regimen include clindamycin for anaerobic coverage.
Pulmonary Edema
Pulmonary edema can result from any disease state that causes increased pulmonary venous pressure or leakiness from pulmonary vasculature, such as:
- Left-sided heart failure
- Myocardial infarction
- ARDS (acute respiratory distress syndrome)
- Valvular disease
- Arrhythmias
Negative pressure pulmonary edema occurs when a patient has upper airway obstruction (eg, laryngospasm during extubation) that results in large negative intrathoracic pressure (due to inspiration against the obstruction), causing noncardiogenic pulmonary edema. It is more common in young men or after head and neck surgery.
Common signs and symptoms of pulmonary edema are caused by difficulties getting enough oxygen to the body, and include:
- Dyspnea
- Orthopnea
- Paroxysmal nocturnal dyspnea
- Wheezing and ronchi
- Dullness to percussion
- Peripheral edema
- S3 or S4 heart sounds
- Tachycardia
When diagnosing pulmonary edema, **increased BNP (brain natriuretic peptide)**or elevated troponin point to a cardiac disease causing the pulmonary edema.
A pulmonary capillary wedge pressure ≥18 mmHg is consistent with cardiogenic cause of pulmonary edema.
QT-prolongation and T-wave inversions are commonly seen on ECG in patients with pulmonary edema of acute onset.
Chest x-ray findings consistent with pulmonary edema include:
- Areas of consolidation
- Kerley B lines (interstitial markings in the lower lungs)
- Cephalization of vessels (increased vessel marking in upper lungs)
The cornerstone of treatment of pulmonary edema is furosemide diuresis. Other adjunctive treatments include supplemental O2 and nitroglycerin (in patients without hypotension).
Inotropic agents such as milrinone, dopamine and dobutamine can be used in patients with severe left ventricular systolic dysfunction and low output syndrome to maintain systemic perfusion pressures.
Empyema is characterized by purulent fluid within the pleural space (between the parietal and visceral pleura).
Empyema is most often a complication of pneumonia due to parapneumonic effusions becoming infected. Therefore the most common infectious causes of pneumonia are also the most common infectious causes of empyema.
Patients with empyema will generally present late in the course of their pneumonia with:
- Worsening fever
- Pleuritic chest pain
- **Decreased fremitus **
- Decreased breath sounds
- **Dullness to percussion **
On CXR, empyemas may cross fissure lines and have a lenticular shape and tapered air-fluid level to the chest wall.
The treatment of choice for empyemas is antibiotics and **drainage via tube thoracostomy. **
Video-assisted thoracoscopic surgery (VATS) should be used to treat empyema that is refractory to treatment with antibiotics and tube thoracostomy or empyema with multiple loculations.
Pleural Effusion

- TB: lymphocytosis
transudate:
- Decreased intrapleural pressure (eg, atelectasis) - reduced perivascular pressure pulls fluid across the vascular membrane into the pleural space
- Decreased plasma oncotic pressure (eg, hypoalbuminemia) - fluid moves into tissue and space (eg, abdominal, pleural) until pressures equalize
- Elevated hydrostatic pressure (eg, congestive heart failure) - increased microvascular pressure pushes fluid through the vascular membrane into the pleural space
PE
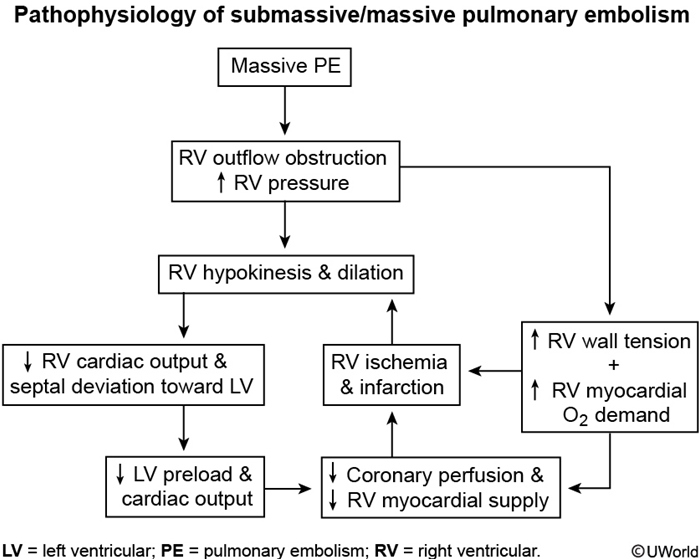
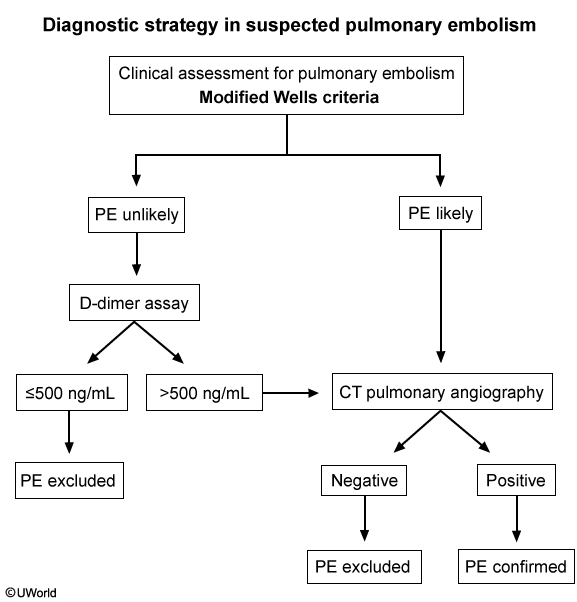
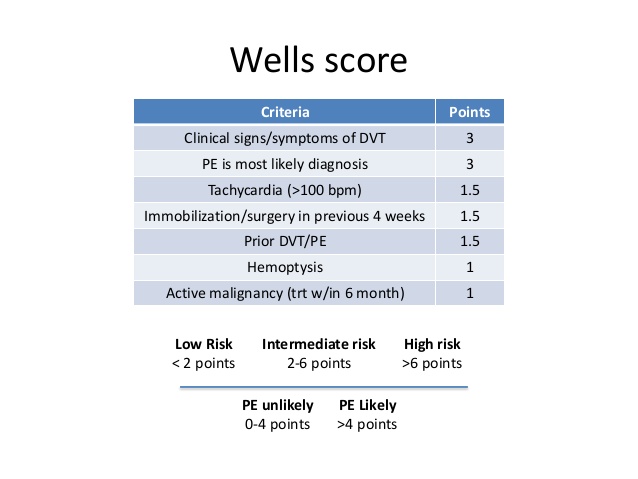
This patient's underlying malignancy (prothrombotic state) and acute presentation of dyspnea, chest pain, tachycardia, hypoxia, and clear lungs are highly suggestive of pulmonary embolism (PE). Patients with acute massive PE can present with syncope and hemodynamic collapse. Examination can show an accentuated (loud) pulmonic component of the second heart sound and elevated central venous pressure (eg, high jugular venous pressure [JVP]).
Acute massive PE results in abrupt increases in pulmonary vascular resistance and, subsequently, right ventricular (RV) pressures. Elevated RV pressures cause increased RV wall tension, cardiac muscle stretching, and RV dilation. This increases RV myocardial oxygen demand and decreases coronary artery perfusion, which causes supply/demand mismatch and RV ischemia. Consequent RV dysfunction can lead to an inability to pump blood through the pulmonary circulation, decreased venous return to the left atrium, decreased cardiac output, and potential hemodynamic collapse.
Bedside echocardiogram can show bowing of the septum into the left ventricle (due to RV pressure exceeding left ventricular diastolic pressure) and RV free-wall hypokinesis with sparing of the apex. The presence of RV dysfunction, along with elevated brain natriuretic peptide and troponin levels, is associated with increased mortality risk.
(It may be difficult to differentiate PE from RV myocardial infarction [MI], which can also cause RV dysfunction; however, RVMI is less likely to cause dyspnea or syncope and more likely to cause bradycardia or arrhythmias).
Aspiration
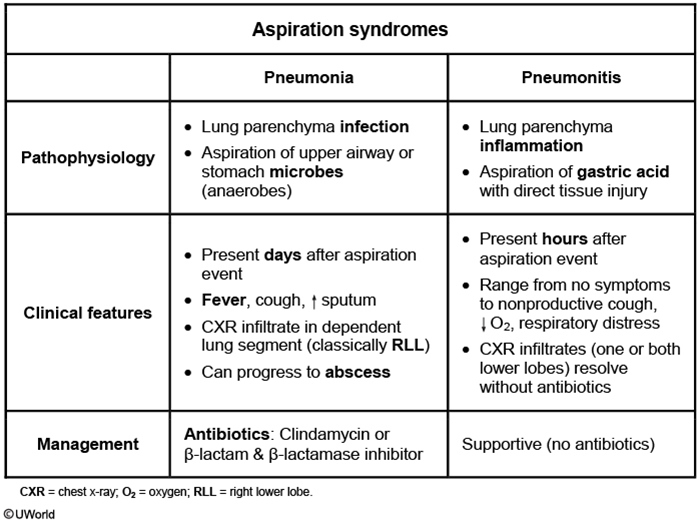

This patient had a witnessed aspiration event (vomiting during intubation that required suctioning) a few hours ago and now has profound hypoxemia with bilateral infiltrates on chest x-ray. He likely has aspiration pneumonitis, an acute lung injury due to aspiration of acidic and sterile stomach contents. The gastric acid induces a chemical burn and consequent inflammatory response, usually within hours of the aspiration event. Bilateral infiltrates can sometimes be seen on chest imaging. The symptoms (and infiltrates) usually resolve rapidly (24-48 hours) with supportive management.
By contrast, aspiration pneumonia (Choice B), which usually occurs in individuals with impaired level of consciousness (eg, following overdose, seizure, or anesthetic use), is caused by inhalation of pathogenic bacteria that colonize oropharyngeal secretions. A typical presentation would be that of an institutionalized elderly patient who has dysphagia and develops fever and cough days (not hours) following an (often unwitnessed) aspiration event. Treatment requires antibiotics.
Patients usually present with indolent symptoms (days to weeks), foul-smelling sputum, and concurrent periodontal disease. Imaging reveals infiltrate in the lower lobes or right middle lobe (aspiration while upright) or the posterior segment of the upper lobes (aspiration while recumbent). The infectious organisms are frequently oral flora (mixed aerobic and anaerobic). Broad-spectrum antibiotics with good anaerobic coverage (eg, clindamycin, amoxicillin-clavulanate) are the mainstay of treatment.
Pulmonary HTN
Pulmonary hypertension (PH) is defined as an elevated pulmonary arterial pressure above 25 mmHg at rest.
If there is no discernible cause, pulmonary hypertension is considered to be primary. Primary pulmonary hypertension is rare and most often occurs in young or middle aged women.
Up to 80% of patients with heritable, or familial, pulmonary hypertension have a mutation in the BMPR2 gene.
Secondary pulmonary hypertension occurs as a complication of a primary medical condition.
Cardiac conditions that are known to cause pulmonary hypertension include:
- Congenital heart disease in patients with L⇒R shunts
- Valvular disease (eg. mitral and aortic stenosis)
- Left heart failure
Patients with recurrent thromboembolic disease can develop pulmonary hypertension.
Pulmonary hypertension secondary to pulmonary disease includes:
- COPD
- Interstitial lung disease
- Obstructive sleep apnea
- Systemic sclerosis
The symptoms of pulmonary hypertension are nonspecific which often causes a delay in diagnosis. Common symptoms include:
- Exertional dyspnea
- Fatigue
- Lethargy
Patients with pulmonary hypertension have their symptoms exaggerated upon exertion.
Auscultation of chest wall in patients with pulmonary hypertension will initially reveal a loud P2 heart sound and right ventricular heave on physical exam.
The initial diagnostic test of choice for patients with suspected pulmonary hypertension is echocardiogram. Findings will include:
- Dilated pulmonary arteries
- Dilated and hypertrophied right atrium and right ventricle
- Abnormal movement of the interventricular septum
A definitive diagnosis requires right heart catheterization demonstrating mean pulmonary artery pressure is ≥25 mmHg at rest.
Chest X-ray classically demonstrates right ventricular hypertrophy and enlargement of the pulmonary arteries.
EKG can demonstrate:
- Right axis deviation
- Peaked P waves (P pulmonale)
- Right ventricular hypertrophy
Treatment for all patients involves addressing the primary medical condition (if there is one) as well as ("SEAD"):
- **Supplemental oxygen **
- Exercise
- Anticoagulation
- Diuretics / Digoxin
Patients with primary pulmonary hypertension due to idiopathic pulmonary arterial hypertension (PAH), heritable PAH, and anorexigen-induced PAH are candidates for _advanced therap_y. The preferred medication is calcium channel blockers in those patients who have been shown to have vasoreactive pulmonary arteries based on testing.
Patients with primary pulmonary hypertension in whom vasoreactive testing is negative can be treated with the following classes of drugs:
- Prostanoids such as epoprostenol, treprostinil and iloprost.
- Endothelin receptor antagonists (-sentan) such as ambrisentan, bosentan and macitentan.
- PDE5 inhibitors such as sildenafil (Viagra).
Lung transplantation is considered the last effective treatment option for patients with idiopathic pulmonary hypertension refractory to medical management.
Cor Pulmonale
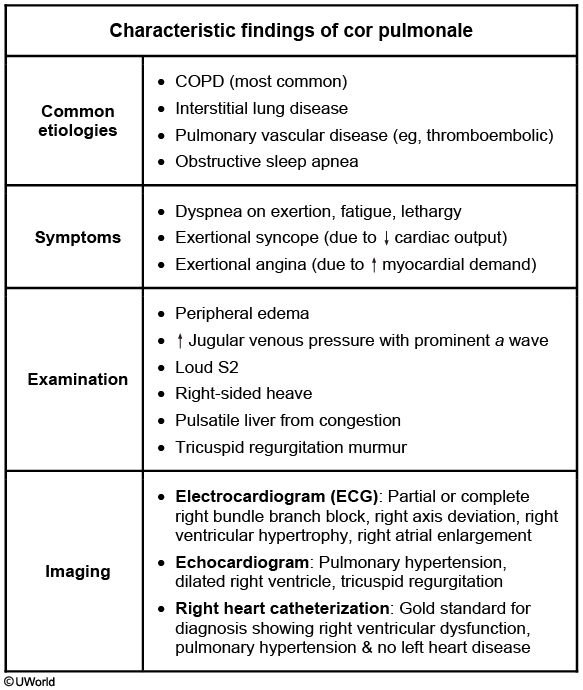
This patient's presentation suggests cor pulmonale, defined as right-sided heart failure (RHF) from pulmonary hypertension (PH). PH may be idiopathic or due to chronic obstructive pulmonary disease (COPD), interstitial lung disease (eg, idiopathic pulmonary fibrosis), obstructive sleep apnea, pulmonary vascular disease (eg, thromboembolic), or chest wall disorders (eg, kyphoscoliosis). RHF due to left-sided or congenital heart disease is not considered cor pulmonale. COPD is the most common cause of cor pulmonale in the United States, with nearly 25% of COPD patients developing this disorder.
Cor pulmonale typically has a gradual onset but can present acutely due to a sudden increase in pulmonary artery pressures (eg, pulmonary embolism). Patients often develop exertional symptoms (eg, dyspnea, angina, syncope). Physical examination may show loud P2 (pulmonic component of the 2nd heart sound), tricuspid regurgitation murmur (holosystolic at the left lower sternal border), elevated jugular venous pressure (JVP), peripheral edema, hepatomegaly due to hepatic congestion, and possible ascites. COPD patients usually have distant heart sounds due to hyperinflated lungs. End-stage cor pulmonale may present with hypotension, tachycardia, and other signs of cardiogenic shock due to decreased stroke volume.
Chest x-ray may show enlarged central pulmonary arteries and loss of retrosternal air space due to right ventricular hypertrophy. Electrocardiogram usually shows right axis deviation, right bundle branch block, right ventricular hypertrophy, and right atrial enlargement. Right heart catheterization is the gold standard for diagnosis and typically shows elevated central venous pressure, right ventricular end-diastolic pressure, and mean pulmonary artery pressure (>25 mm Hg at rest) without left heart disease. Treatment involves optimizing right ventricular dynamics (preload, afterload, and contractility) with supplemental oxygen, diuretics, treatment of underlying etiology, and intravenous inotropes for severe decompensation.
Langerhans Histiocytosis
Pulmonary Langerhans cell histiocytosis (previously histiocytosis X or eosinophilic granuloma) is a rare form of interstitial lung disease that primarily affects young adults.
90% of affected are cigarette smokers.
Langerhans cells express CD1A and **S100 **and possess large cytoplasmic "tennis-racket" Birbeck granules.
Pulmonary Langerhans cell histiocytosis (PLCH) most often presents in a young patient with dyspnea and non-productive cough.
Spontaneous pneumothorax or **diabetes insipidus **are two complications seen in patients with pulmonary Langerhans cell histiocytosis (PLCH).
High-resolution CT is the imaging study of choice in patients with suspected pulmonary Langerhans cell histiocytosis.
The finding of multiple cysts and nodules in the middle to upper lung zones (vs. lower lung zone predominance in idiopathic pulmonary fibrosis) on CT in a young smoker is considered diagnostic for the disease.
Stellate lesions extending into the lung parenchyma is the characteristic histopathologic lesion characteristic of pulmonary Langerhans cell histiocytosis.
Treatment of pulmonary Langerhans cell histiocytosis (PLCH) revolves around smoking cessation as corticosteroids and cytotoxic agents have proven to have limited value.
CF
Cystic fibrosis (CF) is an autosomal recessive inherited disease caused by a mutation in the gene coding for a complex chloride channel. It results in thick, viscous secretions that affect the lungs, pancreas, liver, and reproductive system.
CF is caused by mutations in the gene that codes for the cystic fibrosis transmembrane conductance regulator (CFTR) protein. It is located on chromosome 7.
The most common mutation is delta F508 (deletion of the three bases coding for phenylalanine, which is the 508th amino acid in the CFTR protein).
The CFTR protein normally functions as a cAMP-activated chloride channel on the epithelial cells of the respiratory tract, pancreas, sweat/salivary glands, intestines, and reproductive system.
The defective CFTR channel in CF results in decreased chloride secretion and increased reabsorption of sodium and water by epithelial cells. This leads to dehydration at the cell surface and abnormally thick secretions. These secretions are more difficult to clear, and are more prone to harborinfectious organisms.
Common infection-causing pathogens in the lungs of cystic fibrosis patients include:
- Haemophilus influenzae - common in early childhood
- Staphylococcus aureus - common in early childhood
- Pseudomonas aeruginosa- more prevalent later in life
- Burkholderia cepacia
In the gastrointestinal tract, abnormal bile and pancreatic secretions cause maldigestion and malabsorption, including fat soluble vitamin deficiencies (A, D, E, and K).
More than 95% of men with cystic fibrosis are infertile because of problems with sperm transport, though they are able to produce normal sperm. It is very common in men with CF to have congenital bilateral absence of the vas deferens.
CF is the most common life-shortening autosomal recessive disease in Caucasians, and occurs once in every 2000 to 3000 live births. The disease is most common in the Caucasian population, followed by the Hispanic population.
Symptoms of cystic fibrosis usually begin to present in early childhood and commonly include meconium ileus, respiratory symptoms, and failure to thrive.
Respiratory symptoms of CF may include persistent, productive cough and wheezing. If the disease progresses untreated, patients may experience increased cough, tachypnea, dyspnea, increased sputum production, malaise, anorexia, and weight loss.
The majority of cystic fibrosis patients develop sinus disease, including pan-opacification of the paranasal sinuses, and, less commonly, nasal polyposis.
Symptoms of CF-related pancreatic insufficiency may include steatorrhea and failure to thrive or poor weight gain.
Meconium ileus, distal intestinal obstruction, rectal prolapse, and duodenal ulcers are among the GI manifestations of cystic fibrosis.
The majority of cystic fibrosis cases in the United States are now detected through newborn screening. All infants undergo initial screening, commonly using a serum immunoreactive trypsin (IRT) assay.
Infants with positive newborn screening should undergo sweat chloride testing to confirm the diagnosis. Patients with cystic fibrosis will have elevated sweat chloride > 60 mEq/L. Abnormal sweat chloride testing is sufficient to diagnose CF in infants and is the diagnostic test of choice for non-infant patients.
In adults and older children, two criteria must be met for the diagnosis of CF: Clinical symptoms consistent with CF in at least one organ system AND evidence of CFTR dysfunction (elevated sweat chloride, presence of two disease-causing mutations in CFTR, or abnormal nasal potential difference.)
Cystic fibrosis may be suspected on prenatal ultrasound if there is evidence of meconium peritonitis, bowel dilation, or absent gallbladder.
Findings consistent with cystic fibrosis on physical exam include wheezing, increased anteroposterior diameter of the thorax, digital clubbing, and poor weight gain/failure to thrive.
While chest x-ray is not used for diagnosis, the chest x-rays of cystic fibrosis patients may show hyperinflation, bronchiectasis, cyst formation, and flattening of the diaphragms.
The complications of cystic fibrosis are many and varied. Respiratory complicationsare the major contributors to morbidity and mortality in CF.
Common pulmonary cystic fibrosis complications are hemoptysis (frequent coughing and inflammation leads to the erosion of the walls of bronchial arteries), bronchiectasis, and pneumothorax.
Progressive obstructive lung disease and hypoxia can eventually lead to chronic pulmonary hypertensionand then to right heart failure in CF patients.
Patients may also develop pancreatitis or diabetes due to pancreatic damage caused by thick, abnormal secretion.
Treatment of CF varies based on the specific mutation and severity of disease, but typically includes:
- Short-acting inhaled beta-2 agonists
- Inhalation of hypertonic saline
- Prophylactic antibiotics
- Chest physiotherapy
- Dornase alfa
- A high protein/high calorie diet
- Pancreatic enzyme replacement therapy
Cystic fibrosis lung disease exacerbations are typically treated with aggressive chest physiotherapy and additional antibiotics.
Cystic fibrosis patients with severe disease may be lung transplant candidates.
Sarcoidosis
Sarcoidosis is an immune mediated inflammatory disease of unknown etiology characterized bynoncaseating granulomas in multiple organs, but most notably the lungs.
The incidence of sarcoidosis is significantly higher in young African American females.
The development of sarcoidosis is due to an overreaction by T-cells to an unknown antigen**. CD4 T-helper cells release cytokines** which cause development of non-caseating granulomas.
Noncaseating granulomas consist of macrophages, epithelioid cells, and multinucleated giant cells, surrounded by lymphocytes, monocytes, mast cells and fibroblasts.
Patients with sarcoidosis commonly present between 20 and 60 years of age with:
- Cough
- Dyspnea
- Chest pain
- Eye lesions (e.g. anterior uveitis)
- Skin lesions (e.g. erythema nodosum, lupus pernio)
- Fever
- Anorexia
- Arthralgias
- Parotid gland swelling
Sarcoidosis skin findings include:
- erythema nodosum (tender nodules on the shin)
- **Lupus pernio **(a violaceous rash involving the nose and cheeks)
Anterior uveitis is the most common ocular finding in sarcoidosis.
Features of sarcoidosis are GRUELING:
- Granulomas (noncaseating)
- aRthritis
- Uveitis (anterior)
- Erythema nodosum
- Lymphadenopathy
- Interstitial fibrosis
- Negative TB test
- Gammaglobulinemia
In the lungs, interstitial inflammation leads to fibrosis and ultimately restrictive lung disease.
Patients with a suspicious history for sarcoidosis should receive a chest x-ray.
Chest x-ray is central to the workup of sarcoidosis and reveals bilateral hilar lymphadenopathy and interstitial infiltrates.
Following a suspicious chest x-ray for sarcoidosis (e.g. bilateral hilar lymphadenopathy), a patient should undergo a lymph node biopsy demonstrating non-caseating granulomas.
Lab abnormalities associated with sarcoidosis include:
- Increased serum levels of ACE (angiotensin converting enzyme) and gammaglobulins
- Hypercalcemia and hypercalciuria
- Increased levels of vitamin D (macrophages inside the granulomas convert vitamin D to its active form.)
- Elevated alkaline phosphatase if there is liver involvement
Pulmonary function tests may demonstrate a restrictive pattern, however they can also be normal in some patients.
The most common manifestation of cardiac sarcoidosis are the following (from most common to least):
- Heart block: 3rd degree (most common) or 1st degree
- Ventricular arrhythmia: sustained or nonsustained ventricular tachycardia and ventricular premature beats
Many patients with asymptomatic non-progressive sarcoidosis **do not require treatment and will have spontaneous remission. **
Patients with symptomatic sarcoidosis are managed initially with glucocorticoids such as prednisone (Deltasone)
Patients with symptomatic sarcoidosis that is refractory to glucocorticoids are managed with:
- **Methotrexate **(Trexall)
- **Infliximab **(Remicade)
- Lung transplantation
Sleep Apnea
Sleep apnea is a sleep disorder characterized by repetitive cycles of hypoxia leading to gasping and possibly awakening from sleep. Sleep apnea may be central, obstructive, or mixed in etiology.
Obstructive sleep apnea is defined as a transient obstruction of the upper airway leading to hypoxemia and arousal from sleep.
Risk factors for obstructive sleep apnea include:
- **Obesity **(weight of neck may cause airway compression)
- Anatomical abnormalities (e.g. enlarged tonsils or uvula)
- Family history
- Use of sedatives (e.g. benzodiazepines, alcohol)
- Hypothyroidism
- Smoking
In children, adenotonsillar hypertrophy as well as obesity are major risk factors for obstructive sleep apnea.
Central sleep apnea is characterized by adecrease in ventilatory drive during sleep, leading to episodes of hypoxemia.
Central sleep apnea may be idiopathic(aka primary), or caused secondarily by:
- Stroke
- Arnold-Chiari malformation
- Opioids
- Congestive heart failure (associated with Cheyne-Stokes respiration)
- Any condition affecting the brain stem
Obesity hypoventilation syndrome is defined as the presence of chronic daytime alveolar hypoventilation (PaCO2 >45 mmHg) that occurs in an obese (BMI ≥30 kg/m2) patient and that cannot be attributed to alternative causes of hypercapnia.
Patients with sleep apnea typically present with:
- Daytime somnolence
- Snoring or choking during the night
- Morning headaches
- Poor concentration
- Decreased libido
The diagnosis of sleep apnea is made by polysomnography.
The first line treatment for obstructive sleep apnea includes both weight loss and continuous positive airway pressure (CPAP).
Patients with obstructive sleep apnea refractory to continuous positive airway pressure (CPAP) and weight loss may be treated with:
- Uvulopalatopharyngoplasty (removal of excess oropharyngeal tissues)
- Tracheostomy (last resort)
Central sleep apnea is treated with:
- CPAP (if hyperventilation present)
- BiPAP (if hypoventilation present)
- Supplemental oxygen
- Acetazolamide (Diamox)
If left untreated, patients with sleep apnea may develop:
- Impaired alertness leading to motor vehicle collisions
- Pulmonary hypertension(may lead to cor pulmonale)
- Systemic hypertension
- Arrhythmias
- Polycythemia
Bronchogenic Cyst
The diagnosis of mediastinal tumors is based on chest-x rays and CT scans. A bronchogenic cyst may be seen on the AP chest x-ray. The diagnosis is best made with a CT scan. Bronchogenic cysts are located in the middle mediastinum and are benign entities. Other middle mediastinal masses include: tracheal tumors, pericardial cysts, lymphoma, lymph node enlargement, and aortic aneurysms of the arch
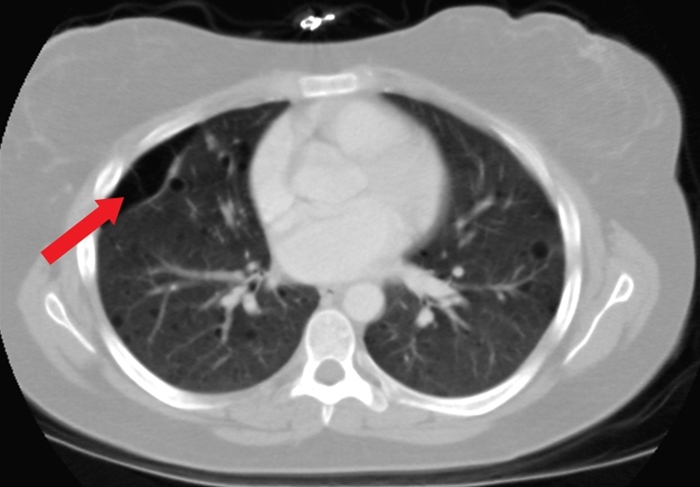
Secondary Spontaneous Pneumo
This patient with chronic obstructive pulmonary disease (COPD) and acute-onset shortness of breath, hypoxia, and unilaterally decreased breath sounds likely has a secondary spontaneous pneumothorax (SSP). SSP occurs in patients with known lung disease (eg, COPD, cystic fibrosis). Other common manifestations include chest pain and hyper-resonance on percussion. Cigarette smoking markedly increases the risk of pneumothorax. Chronic destruction of alveolar sacs leads to the formation of large alveolar blebs (red arrow), which can eventually rupture and leak air into the pleural space.